




Evaluación funcional en un paciente con miopatía congénita centronuclear asociada a Diamina 2
La miopatía centronuclear es una enfermedad congénita que debe su nombre a la localización central del núcleo asociada a una hipotrofia de las fibras musculares, en especial las de tipo 1. De acuerdo al modo de herencia y a la presentación clínica se reconocen 3 tipos principales: Ligada a X – (MTM1), autosómica recesiva (anfifisina 2) y autosómica dominante (dinamina 2 – DNM2).1 El gen de la DNM2 se encuentra en el cromosoma 19p13.2 y codifica una GTPasa que se encarga de la endocitosis, así mismo, esta proteína se relaciona con el transporte intracelular y la organización del citoesqueleto.2 Se han encontrado al menos 20 mutaciones genéticas.2 El gen de la DNM2 también se ha relacionado con el Charcot Marie Tooth.1
El espectro clínico de la miopatía centronuclear es muy amplio. Las características fenotípicas más frecuentes son la debilidad de predominio distal, en especial de los músculos dorsiflexores, la oftalmoplejia y la ptosis palpebral. Usualmente la debilidad muscular es leve y a veces se detecta en la edad adulta. La progresión es lenta, conservando al menos la fuerza antigravitatoria. Hacia la quinta a sexta década de la vida, estos pacientes requieren trasladarse en silla de ruedas. El déficit cognitivo y las alteraciones cardiopulmonares no son frecuentes.3 4 Sin embargo, en la variante específica de la dinamina 2, el fenotipo característico suele comprometer severamente la función motora gruesa, siguiendo un curso rápidamente progresivo y altamente limitante. 5
En el siguiente texto presentamos el caso de un paciente con miopatía centronuclear por alteración de la dinamina 2, destacando los cambios funcionales, los cuales fueron evaluados a través de pruebas específicas y escalas de función motora.
Caso clínico
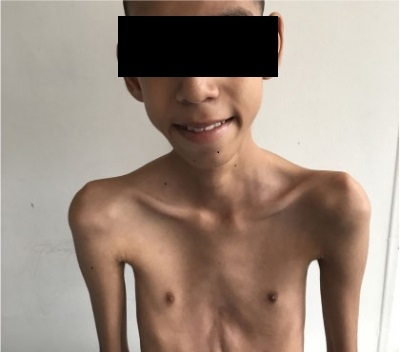
Se trata de un adolescente colombiano de 15 años, con diagnóstico de miopatía centronuclear de carácter autosómico dominante por alteración en el gen DNM2, variante patogénica c. 1856 c>T (p, Ser619Leu). Este paciente presentó hipoxia neonatal y requirió ventilación invasiva al momento del nacimiento, con necesidad de oxígeno suplementario hasta el año y medio de edad, así mismo, se documentó un retraso en el desarrollo motor, asociado a debilidad e hipotrofia muscular generalizada.
En los estudios paraclínicos se encontraron niveles de fosfocreatinin quinasa (CPK) ligeramente elevados (311 mg/dl). La biopsia muscular evidenció atrofia muscular con reemplazo graso, sin actividad inflamatoria asociada. Se encontró en el estudio de cinedeglución una disfagia en todas las fases secundaria a debilidad muscular y en la polisomnografía un SAHOS leve. La resonancia cerebral simple y el ecocardiograma fueron normales.
Las pruebas funcionales (Tabla 1), mostraron una mejoría significativa en la función motora gruesa y en las destrezas manuales hasta los 11 años de edad. Posteriormente, el niño presentó un deterioro funcional de rápida evolución. A los 13 años de edad perdió la capacidad de marcha independiente y se documentaron deformidades en flexión de caderas y rodillas asociado a una debilidad muscular generalizada, con requerimiento de silla de ruedas para los traslados.
Actualmente la fuerza muscular en cintura escapular y pélvica es de 3+/5, en músculos distales de miembros superiores de 3/5 y en inferiores de 0/5, asociado a arreflexia generalizada.
| MFM | 9 años | 10 años | 11 años | 14 años |
|---|---|---|---|---|
| Bipedestación | 48,718 | 51,282 | 53,846 | 15,385 |
| Motora axial | 88,889 | 91,667 | 91,667 | 86,111 |
| Motora distal | 71,429 | 80,952 | 76,190 | 80,952 |
| Up and Go | 14,96 | 9,5 | 12 | NA |
| Vignos | 2 | 2 | 3 | 9 |
| Brooke | 1 | 1 | 1 | 2 |
otra tabla
| 12 | 3 | 4 |
| 42 | 5 | 6 |
Discusión
Este caso muestra los resultados de las pruebas funcionales en un paciente adolescente con miopatía centronuclear asociado a la alteración de la dinamina 2, variante patogénica puntual en el gen DMN2 [c. 1856 c>T (p, Ser619Leu)]. Esta variante ha sido previamente descrita en la literatura en 11 familias distribuidas en el mundo entero (Bélgica, Canadá, USA, Argentina).5 En todos los casos descritos, al igual que en este paciente, se presentó un patrón de inicio neonatal caracterizado por ptosis palpebral, oftalmoplejía, hipotonía generalizada severa, Apgar bajo y requerimiento de ventilación mecánica.5 Aunque la mayoría de casos suelen cursar con un retraso severo del neurodesarrollo con imposibilidad para lograr la marcha, nuestro paciente alcanzó dicho hito, presentado en la etapa de la adolescencia un franco deterioro en la función motora gruesa.
Para el seguimiento de este paciente, se aplicaron escalas convencionales de funciones corporales (Medical Research Council -MRC) y escalas de actividades (Medida de la Función Motora-MFM, prueba de caja y cubos). Estas han sido utilizadas en pacientes con diferentes enfermedades neuromusculares6-9.
Las medidas de la fuerza muscular pueden ser difíciles de aplicar en pacientes pediátricos y dada la amplia gama de enfermedades con diferentes patrones de afectación muscular, dicha evaluación se torna compleja para el examinador.7 Por lo anterior, escalas como la MFM son de utilidad para monitorizar la severidad y la progresión de enfermedades con curso gradual9. Incluso se ha propuesto una escala modificada mediante análisis Rash específica para miopatías congenitas10.
En nuestro paciente, se encontró mejoría en las primeras evaluaciones funcionales, con un deterioro abrupto entre los 11 y los 14 años, comprometiendo los puntajes de la escala de Vignos y Brooke, el dominio 1 (bipedestación) de la escala MFM y en la prueba de caja y cubos, lo cual corresponde a un compromiso de las habilidades relacionadas con la marcha y de las destrezas manuales.
En la literatura actual, la descripción de la evaluación funcional en estos pacientes es limitada y los seguimientos son por cortos períodos. Ningún estudio publicado hasta el presente, posee una evaluación con datos específicos en la función motora gruesa y fina. En nuestro paciente se logró documentar los cambios funcionales durante un periodo de 6 años.
Si bien, las miopatías congénitas tienen un amplio espectro clínico, el hecho de objetivar los cambios en la evolución de cualquier paciente debe ser un pilar en todo proceso de rehabilitación. Esto permite establecer un pronóstico asertivo desde el punto de vista funcional, instaurar metas de tratamiento y discernir apropiadamente sobre las diferentes opciones terapéuticas en un momento dado, según el curso de la enfermedad.
Es probable que factores externos como el acceso oportuno a programas de rehabilitación, las condiciones geográficas y socioeconómicas del paciente, las características propias del sistema de salud y la disponibilidad de ayudas diagnósticas avanzadas, pudieran haber influido en el desenlace funcional de este paciente.11 Sin embargo, el registro y seguimiento mediante pruebas funcionales permitió tomar decisiones puntuales que impactaron en la calidad de vida.
A futuro, los equipos de atención deberían enfocar sus esfuerzos en prevenir complicaciones asociadas al inmovilismo relativo y evitar las principales causas de morbimortalidad de los pacientes con miopatías congénitas,12 como lo son las enfermedades pulmonares y cardiovasculares. El abordaje debe ser interdisciplinario y a través del seguimiento de habilidades funcionales que faciliten tomar decisiones en la conducta terapéutica y en los procesos inherentes a su rehabilitación.
Si bien, las miopatías congénitas tienen un amplio espectro clínico, el hecho de objetivar los cambios en la evolución de cualquier paciente debe ser un pilar en todo proceso de rehabilitación.
Autores
Referencias
- Aghbolaghi AG, Lechpammer M. A rare case of centronuclear myopathy with DNM2 mutation: genotype-phenotype correlation. Autops Case Rep 2017;7(2):43-48. doi: 10.4322/acr.2017.020 [published Online First: 2017/06/30]
- Trochet D, Prudhon B, Jollet A, et al. Reprogramming the Dynamin 2 mRNA by Spliceosome-mediated RNA Trans-splicing. Mol Ther Nucleic Acids 2016;5(9):e362. doi: 10.1038/mtna.2016.67 [published Online First: 2016/09/13]
- Verma S, Balasubramanian SB. Clinical, Electrophysiology, and Pathology Features of Dynamin Centronuclear Myopathy: A Case Report and Review of Literature. J Clin Neuromuscul Dis 2016;18(2):84-88. doi: 10.1097/CND.0000000000000141
- Witting N, Werlauff U, Duno M, et al. Phenotypes, genotypes, and prevalence of congenital myopathies older than 5 years in Denmark. Neurol Genet 2017;3(2):e140. doi: 10.1212/NXG.0000000000000140 [published Online First: 2017/03/21]
- Böhm J, Biancalana V, Dechene ET, et al. Mutation spectrum in the large GTPase dynamin 2, and genotype-phenotype correlation in autosomal dominant centronuclear myopathy. Hum Mutat 2012;33(6):949-59. doi: 10.1002/humu.22067 [published Online First: 2012/04/04]
- Florence JM, Pandya S, King WM, et al. Intrarater reliability of manual muscle test (Medical Research Council scale) grades in Duchenne’s muscular dystrophy. Phys Ther 1992;72(2):115-22; discussion 22-6.
- Escolar DM, Henricson EK, Mayhew J, et al. Clinical evaluator reliability for quantitative and manual muscle testing measures of strength in children. Muscle Nerve 2001;24(6):787-93.
- Clark R, Locke M, Hill B, et al. Clinimetric properties of lower limb neurological impairment tests for children and young people with a neurological condition: A systematic review. PLoS One 2017;12(7):e0180031. doi: 10.1371/journal.pone.0180031 [published Online First: 2017/07/03]
- de Lattre C, Payan C, Vuillerot C, et al. Motor function measure: validation of a short form for young children with neuromuscular diseases. Arch Phys Med Rehabil 2013;94(11):2218-26. doi: 10.1016/j.apmr.2013.04.001 [published Online First: 2013/04/18]
- Vuillerot C, Rippert P, Kinet V, et al. Rasch analysis of the motor function measure in patients with congenital muscle dystrophy and congenital myopathy. Arch Phys Med Rehabil 2014;95(11):2086-95. doi: 10.1016/j.apmr.2014.06.005 [published Online First: 2014/06/25]
- Ruiz-Cortes X, Ortiz-Corredor F, Mendoza-Pulido C. Reliability of home-based, motor function measure in hereditary neuromuscular diseases. J Int Med Res 2017;45(1):261-71. doi: 10.1177/0300060516674608 [published Online First: 2017/01/12]